News & Announcements
Read the latest news and announcements from Northwestern University Feinberg School of Medicine’s Galter Health Sciences Library & Learning Center.
-
 06.12.2026The Galter Library is constantly striving to update our electronic resources and bring current tools to our patrons' desktops.
06.12.2026The Galter Library is constantly striving to update our electronic resources and bring current tools to our patrons' desktops. -
 06.05.2026The poster reports on ongoing research examining librarian influence on the quality of systematic reviews published by Feinberg School of Medicine authors.
06.05.2026The poster reports on ongoing research examining librarian influence on the quality of systematic reviews published by Feinberg School of Medicine authors. -
 06.01.2026The Big Ten Academic Alliance (BTAA) Center for Library Programs has secured an Open Publishing Agreement with Wiley through December 31, 2027
06.01.2026The Big Ten Academic Alliance (BTAA) Center for Library Programs has secured an Open Publishing Agreement with Wiley through December 31, 2027 -
 05.18.2026Commentary: Onus is on researchers to use GenAI responsibly and ensure integrity, proper attribution
05.18.2026Commentary: Onus is on researchers to use GenAI responsibly and ensure integrity, proper attribution -
 05.18.2026The Galter Library is constantly striving to update our electronic resources and bring current tools to our patrons' desktops.
05.18.2026The Galter Library is constantly striving to update our electronic resources and bring current tools to our patrons' desktops. -
 05.15.2026A new Galter Library search interface offers a more streamlined and accessible search experience.
05.15.2026A new Galter Library search interface offers a more streamlined and accessible search experience. -
 05.13.2026James S. T. Yao, MD, PhD, was a leading vascular surgeon who made an indelible impact on the field at Northwestern Medicine.
05.13.2026James S. T. Yao, MD, PhD, was a leading vascular surgeon who made an indelible impact on the field at Northwestern Medicine. -
 05.05.2026Kristi Holmes, PhD, associate dean for knowledge management and strategy and director of the Galter Health Sciences Library and Learning Center, has been named the 2026 Medical Librarian of the Year by the Friends of the National Library of Medicine.
05.05.2026Kristi Holmes, PhD, associate dean for knowledge management and strategy and director of the Galter Health Sciences Library and Learning Center, has been named the 2026 Medical Librarian of the Year by the Friends of the National Library of Medicine. -
 05.04.2026A new digital archive developed by Northwestern scientists reveals how state-supported research funding agencies cooperate with the scientific community to decide to support scientific research projects and contribute to scientific innovation.
05.04.2026A new digital archive developed by Northwestern scientists reveals how state-supported research funding agencies cooperate with the scientific community to decide to support scientific research projects and contribute to scientific innovation. -
 04.23.2026As the medical director of the Obstetrics and Gynecology Ultrasound Center, Sabbagha quickly became a leader in the field of ultrasound in high-risk obstetrics.
04.23.2026As the medical director of the Obstetrics and Gynecology Ultrasound Center, Sabbagha quickly became a leader in the field of ultrasound in high-risk obstetrics. -
04.10.2026The Galter Library is constantly striving to update our electronic resources and bring current tools to our patrons' desktops.
-
 03.25.2026Lydia Anna Ballard, a pioneering physician and Northwestern‑affiliated alumna, broke barriers in 19th‑century medicine while advancing care for women.
03.25.2026Lydia Anna Ballard, a pioneering physician and Northwestern‑affiliated alumna, broke barriers in 19th‑century medicine while advancing care for women. -
 03.21.2026Publishing research open access increases visibility, expands readership, and helps ensure scholarly work is accessible.
03.21.2026Publishing research open access increases visibility, expands readership, and helps ensure scholarly work is accessible. -
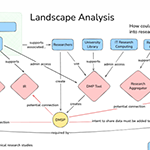 03.20.2026To promote open access to data, research infrastructure and reproducibility, data management and sharing plans (DMSPs) are being used at academic institutions.
03.20.2026To promote open access to data, research infrastructure and reproducibility, data management and sharing plans (DMSPs) are being used at academic institutions. -
03.03.2026The Galter Library is constantly striving to update our electronic resources and bring current tools to our patrons' desktops.
-
 02.20.2026The papers of Ralph S. Paffenbarger, Jr., MD, DPH, a Feinberg alumnus and pioneering epidemiologist, are now fully processed and open to researchers.
02.20.2026The papers of Ralph S. Paffenbarger, Jr., MD, DPH, a Feinberg alumnus and pioneering epidemiologist, are now fully processed and open to researchers. -
02.16.2026The Galter Library is constantly striving to update our electronic resources and bring current tools to our patrons' desktops.
-
 02.11.2026James P. Carter, MD, DPH, a 1957 graduate of the Northwestern University Medical School, became a leading and sought-after expert in medicine, especially nutrition.
02.11.2026James P. Carter, MD, DPH, a 1957 graduate of the Northwestern University Medical School, became a leading and sought-after expert in medicine, especially nutrition. -
 02.06.2026Galter Library will host the traveling exhibition, "Care & Custody: Past Responses to Mental Health," in the library atrium from February 9 – March 21, 2026.
02.06.2026Galter Library will host the traveling exhibition, "Care & Custody: Past Responses to Mental Health," in the library atrium from February 9 – March 21, 2026. -
 10.31.2025Feinberg Founders & Notables: Historical Walking Tour of Graceland Cemetery
10.31.2025Feinberg Founders & Notables: Historical Walking Tour of Graceland Cemetery -
 10.09.2025The shift to the Common Forms is a part of the larger NIH initiative to strengthen compliance with federal policy, streamline grant management, and ensure researchers receive proper visibility and credit for their work.
10.09.2025The shift to the Common Forms is a part of the larger NIH initiative to strengthen compliance with federal policy, streamline grant management, and ensure researchers receive proper visibility and credit for their work. -
09.19.2025NIH policy effective July 1, 2025 requires immediate public access to NIH-funded manuscripts. Below, we address our most-frequently asked questions and summarize key updates.
-
 09.04.2025Northwestern alum, William S. Kroger, MD pioneered the field of clinical hypnosis, which he helped legitimize over his 60-year career.
09.04.2025Northwestern alum, William S. Kroger, MD pioneered the field of clinical hypnosis, which he helped legitimize over his 60-year career. -
 09.03.2025Shaw will serve as the library’s lead on policies, mandates, and best practices affecting funded research in the health sciences.
09.03.2025Shaw will serve as the library’s lead on policies, mandates, and best practices affecting funded research in the health sciences. -
07.10.2025View titles that were added in Summer 2025 to subscribed collections licensed by the Library or now available to the Northwestern University community.